
Bêta-thalassémie majeure : Comprendre cette maladie génétique
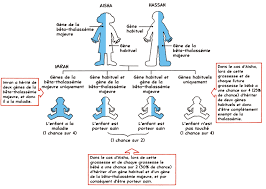
La bêta-thalassémie majeure est une maladie génétique rare mais grave qui affecte la production d’hémoglobine, une protéine essentielle des globules rouges. Cette condition héréditaire se caractérise par une diminution importante de la production de l’hémoglobine bêta, ce qui entraîne une anémie sévère chez les patients atteints.
Les symptômes de la bêta-thalassémie majeure peuvent varier en fonction de la gravité de la maladie, mais ils incluent généralement une fatigue extrême, des problèmes cardiaques, une croissance retardée et des complications osseuses. Les patients atteints de cette forme sévère de thalassémie nécessitent souvent des transfusions sanguines régulières pour compenser le manque d’hémoglobine.
Le traitement de la bêta-thalassémie majeure repose sur la gestion des symptômes et des complications associées à la maladie. Outre les transfusions sanguines, certains patients peuvent bénéficier d’une greffe de moelle osseuse pour remplacer les cellules souches défectueuses responsables de la production d’hémoglobine altérée.
Il est essentiel que les patients atteints de bêta-thalassémie majeure reçoivent un suivi médical régulier et un soutien approprié pour gérer leur condition. La sensibilisation à cette maladie rare est également cruciale pour encourager la recherche et le développement de nouvelles thérapies qui pourraient améliorer la qualité de vie des personnes touchées.
En conclusion, la bêta-thalassémie majeure est une maladie génétique complexe qui nécessite une prise en charge multidisciplinaire pour assurer le bien-être des patients. Avec un diagnostic précoce, un traitement approprié et un suivi médical attentif, il est possible d’améliorer la qualité de vie des personnes vivant avec cette condition rare mais significative.
Questions Fréquemment Posées sur la Bêta-Thalassémie Majeure
- Qu’est-ce que la bêta-thalassémie majeure?
- Quels sont les symptômes de la bêta-thalassémie majeure?
- Comment diagnostique-t-on la bêta-thalassémie majeure?
- Quels sont les traitements disponibles pour la bêta-thalassémie majeure?
- Quelles sont les complications possibles de la bêta-thalassémie majeure?
- Y a-t-il des facteurs de risque associés à la bêta-thalassémie majeure?
- Existe-t-il des mesures préventives pour réduire les complications liées à la bêta-thalassémie majeure?
Qu’est-ce que la bêta-thalassémie majeure?
La bêta-thalassémie majeure est une forme sévère de thalassémie, une maladie génétique affectant la production d’hémoglobine dans le sang. Les personnes atteintes de bêta-thalassémie majeure présentent un déficit important en hémoglobine bêta, ce qui entraîne une anémie sévère et des complications médicales potentiellement graves. Cette condition nécessite souvent un traitement régulier par transfusions sanguines pour pallier le manque d’hémoglobine et maintenir un état de santé optimal chez les patients concernés.
Quels sont les symptômes de la bêta-thalassémie majeure?
La bêta-thalassémie majeure se caractérise par une variété de symptômes qui peuvent varier en gravité d’un individu à l’autre. Les symptômes les plus courants de cette forme sévère de thalassémie incluent une fatigue extrême, une pâleur de la peau, des vertiges, des maux de tête fréquents, une croissance retardée chez les enfants, une augmentation de la taille de la rate et du foie, ainsi que des complications osseuses telles que des fractures et des déformations. Il est essentiel de consulter un professionnel de la santé si l’on suspecte la présence de ces symptômes afin d’obtenir un diagnostic précis et un plan de traitement approprié pour gérer la bêta-thalassémie majeure.
Comment diagnostique-t-on la bêta-thalassémie majeure?
Le diagnostic de la bêta-thalassémie majeure repose généralement sur une combinaison d’analyses sanguines et de tests génétiques. Les médecins peuvent effectuer des tests pour évaluer le taux d’hémoglobine, la taille et la forme des globules rouges, ainsi que la quantité de globules rouges dans le sang. De plus, des analyses génétiques peuvent être réalisées pour identifier les mutations spécifiques responsables de la bêta-thalassémie. Un diagnostic précis est essentiel pour déterminer le traitement approprié et mettre en place une prise en charge adaptée pour les patients atteints de cette maladie génétique rare mais significative.
Quels sont les traitements disponibles pour la bêta-thalassémie majeure?
Pour la bêta-thalassémie majeure, les traitements disponibles visent à gérer les symptômes et les complications de la maladie. Les principales approches thérapeutiques incluent les transfusions sanguines régulières pour compenser le manque d’hémoglobine, ainsi que la chélation du fer pour prévenir l’accumulation excessive de fer dans l’organisme due aux transfusions répétées. Dans certains cas, une greffe de moelle osseuse peut être envisagée comme traitement curatif pour remplacer les cellules souches défectueuses responsables de la production d’hémoglobine altérée. Une approche multidisciplinaire impliquant des médecins spécialisés, des hématologues et d’autres professionnels de la santé est essentielle pour assurer une prise en charge optimale des patients atteints de bêta-thalassémie majeure.
Quelles sont les complications possibles de la bêta-thalassémie majeure?
Les complications possibles de la bêta-thalassémie majeure sont variées et peuvent avoir un impact significatif sur la santé des patients. Parmi les complications les plus courantes de cette forme sévère de thalassémie, on retrouve l’anémie sévère, les problèmes cardiaques liés à la surcharge en fer due aux transfusions sanguines régulières, la croissance retardée chez les enfants, ainsi que des complications osseuses telles que l’ostéoporose. La gestion adéquate de ces complications nécessite une approche médicale spécialisée et un suivi régulier pour assurer le bien-être global des patients atteints de bêta-thalassémie majeure.
Y a-t-il des facteurs de risque associés à la bêta-thalassémie majeure?
La bêta-thalassémie majeure est une maladie génétique héréditaire, ce qui signifie que les principaux facteurs de risque associés à cette condition sont liés à la transmission des gènes défectueux des parents aux enfants. Les individus ayant des antécédents familiaux de thalassémie ont un risque plus élevé de développer la forme majeure de la maladie. De plus, dans certaines populations où la thalassémie est plus fréquente, comme dans les régions méditerranéennes, le Moyen-Orient et l’Asie du Sud-Est, le risque d’hériter de la bêta-thalassémie majeure est plus élevé. Il est donc essentiel pour les couples présentant un risque génétique de consulter un conseiller génétique pour évaluer les probabilités de transmission de la maladie à leur progéniture.
Existe-t-il des mesures préventives pour réduire les complications liées à la bêta-thalassémie majeure?
Il n’existe pas de mesures préventives spécifiques pour réduire les complications liées à la bêta-thalassémie majeure, car cette maladie génétique est inhérente et ne peut être évitée. Cependant, une gestion proactive de la condition est essentielle pour minimiser les effets négatifs sur la santé des patients. Un suivi médical régulier, des transfusions sanguines appropriées, une supplémentation en fer contrôlée et une éducation sur la nutrition et le mode de vie adaptés peuvent contribuer à réduire les risques de complications associées à la bêta-thalassémie majeure. Il est crucial que les patients travaillent en étroite collaboration avec leur équipe médicale pour élaborer un plan de traitement personnalisé visant à optimiser leur qualité de vie malgré les défis posés par cette maladie.